Abstract
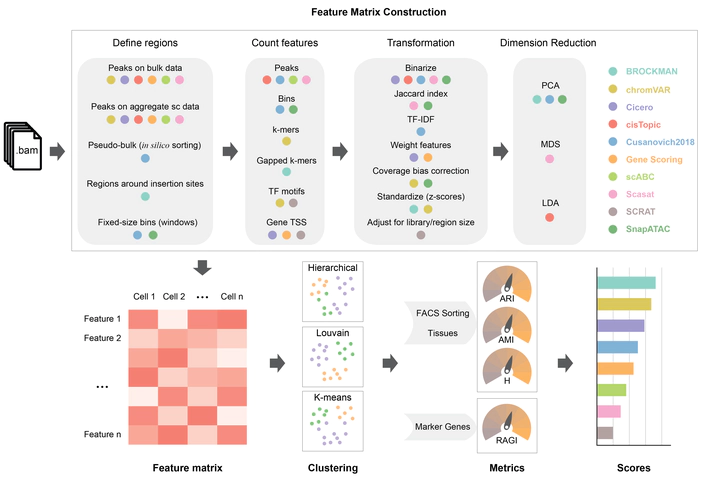
We present a benchmarking framework that is applied to 10 computational methods for scATAC-seq on 13 synthetic and real datasets from different assays, profiling cell types from diverse tissues and organisms. Methods for processing and featurizing scATAC-seq data were compared by their ability to discriminate cell types when combined with common unsupervised clustering approaches. We rank evaluated methods and discuss computational challenges associated with scATAC-seq analysis including inherently sparse data, determination of features, peak calling, the effects of sequencing coverage and noise, and clustering performance. Running times and memory requirements are also discussed.
Type
Publication
Genome Biology