 SIMBA
SIMBA
Abstract
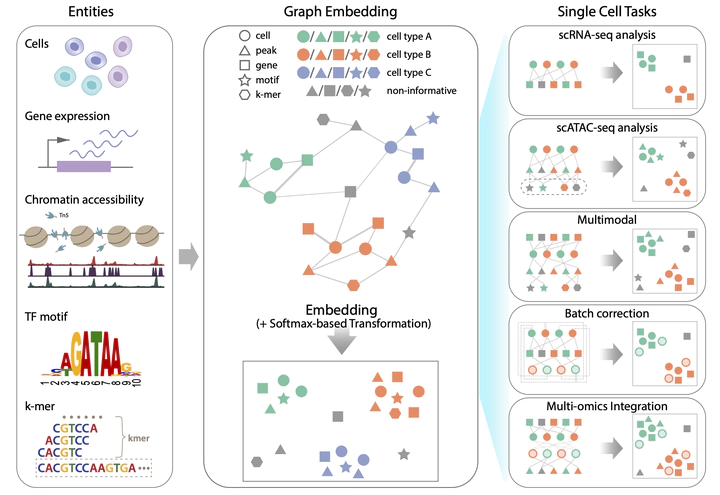
Most current single-cell analysis pipelines are limited to cell embeddings and rely heavily on clustering, while lacking the ability to explicitly model interactions between different feature types. Furthermore, these methods are tailored to specific tasks, as distinct single-cell problems are formulated differently. To address these shortcomings, here we present SIMBA, a graph embedding method that jointly embeds single cells and their defining features, such as genes, chromatin-accessible regions and DNA sequences, into a common latent space. By leveraging the co-embedding of cells and features, SIMBA allows for the study of cellular heterogeneity, clustering-free marker discovery, gene regulation inference, batch effect removal and omics data integration. We show that SIMBA provides a single framework that allows diverse single-cell problems to be formulated in a unified way and thus simplifies the development of new analyses and extension to new single-cell modalities. SIMBA is implemented as a comprehensive Python library (https://simba-bio.readthedocs.io).